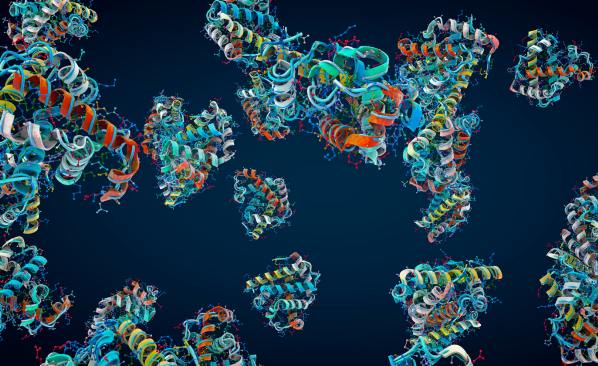
Les protéines, des macromolécules biologiques présentes dans toutes les cellules vivantes, sont formées d’une ou plusieurs chaînes constituées de séquences d’acides aminés. La plupart d’entre elles ont une structure tridimensionnelle complexe qui conditionne leurs propriétés. Elles catalysent des réactions chimiques, renforcent les tissus du corps humain (c’est le rôle du collagène), jouent un rôle important dans le fonctionnement du système immunitaire ; la myoglobine, une protéine du muscle cardiaque, stocke l’oxygène, etc. Elles peuvent aussi enrober la molécule d’un médicament qui doit pénétrer dans une cellule humaine ; des familles de nouvelles protéines sont donc appelées à jouer un rôle croissant en pharmacologie, pour des usages thérapeutiques, pour produire des biomatériaux, etc. Les biologistes étaient d’ailleurs convaincus, de longue date, que la détermination de la structure des protéines était une étape préliminaire et incontournable dans les recherches en génétique et en pharmacologie.
En 2021, une véritable rupture technique est intervenue pour leur prêter main forte. En effet, l’intelligence artificielle utilisant la technique de l’apprentissage profond a permis de « prévoir » très rapidement la structure tridimensionnelle de molécules, notamment de protéines, à partir de la connaissance primaire de séquences de résidus d’acides aminés qui la constituent (donc un nombre limité de ces séquences) [1].
Cette méthode a été mise au point par la société DeepMind, une filiale de Google, connue pour son algorithme du jeu de go AlphaGo. Elle a conçu l’algorithme AlphaFold, utilisant l’intelligence artificielle avec la technique de l’apprentissage profond et, après deux années de travail, en collaboration avec l’Institut de bio-informatique de l’EMBL (European Molecular Biology Laboratory) à Cambridge, elle a déterminé, en 2022, la structure tridimensionnelle de 200 millions de protéines. Celles-ci proviennent d’un très grand nombre d’organismes dont des bactéries (et 98 % des protéines d’origine humaine), constituant ainsi une base de données à laquelle ont accès tous les chercheurs. Les chercheurs de DeepMind ont utilisé une base de données initiale de 200 000 protéines dont la composition et la structure étaient connues et qui leur a permis de réaliser « l’apprentissage » de l’algorithme.
La conformation spatiale des protéines dépend de la nature des acides aminés qui les constituent et de leurs interactions physico-chimiques. Connaissant un nombre limité de ces constituants, l’algorithme « prévoit » rapidement la structure et la conformation d’une protéine à partir de la base de données initiale. Ces prévisions sont loin d’être parfaites (elles résultent d’un calcul statistique) et la structure des molécules doit être vérifiée expérimentalement (par les techniques de cristallographie aux rayons X et de la cryo-microscopie électronique [2]). Selon l’EMBL, 80 % des prévisions seraient soit totalement exactes (35 %), soit suffisamment précises pour être utilisées dans des applications (45 %). La détermination de la forme de protéines et la prévision de leur transformation éventuelle au cours du temps sont des étapes importantes pour envisager des applications de ces molécules.
Cette innovation a donné un coup d’accélérateur aux recherches sur :
- le rôle de protéines dans certaines maladies (elles peuvent favoriser la multiplication de cellules cancéreuses) ;
- de nouvelles familles de protéines utilisables en pharmacologie ;
- la synthèse de nouveaux catalyseurs pour...